


 Sign Out
Sign Out

Pharmacology: Pharmacodynamics: Mechanism of action: Fluticasone furoate/umeclidinium/vilanterol is a combination of inhaled synthetic corticosteroid, long-acting muscarinic receptor antagonist and long-acting beta2-adrenergic agonist (ICS/LAMA/LABA). Following oral inhalation, umeclidinium and vilanterol act locally on airways to produce bronchodilation by separate mechanisms and fluticasone furoate reduces inflammation.
Fluticasone furoate: Fluticasone furoate is a corticosteroid with potent anti-inflammatory activity. The precise mechanism through which fluticasone furoate affects COPD symptoms is not known. Corticosteroids have been shown to have a wide range of actions on multiple cell types (e.g. eosinophils, macrophages, lymphocytes) and mediators (e.g. cytokines and chemokines) involved in inflammation.
Umeclidinium: Umeclidinium is a long-acting muscarinic receptor antagonist (also referred to as an anticholinergic). Umeclidinium exerts its bronchodilatory activity by competitively inhibiting the binding of acetylcholine with muscarinic receptors on airway smooth muscle. It demonstrates slow reversibility at the human M3 muscarinic receptor subtype in vitro and a long duration of action in vivo when administered directly to the lungs in pre-clinical models.
Vilanterol: Vilanterol is a selective long-acting, beta2-adrenergic receptor agonist (LABA). The pharmacologic effects of beta2-adrenergic agonists, including vilanterol, are at least in part attributable to stimulation of intracellular adenylate cyclase, the enzyme that catalyses the conversion of adenosine triphosphate (ATP) to cyclic-3',5'- adenosine monophosphate (cyclic AMP). Increased cyclic AMP levels cause relaxation of bronchial smooth muscle and inhibition of release of mediators of immediate hypersensitivity from cells, especially from mast cells.
Pharmacodynamic effects: Cardiac electrophysiology: The effect of fluticasone furoate/umeclidinium/vilanterol on the QT interval has not been evaluated in a thorough QT (TQT) study. TQT studies for FF/VI and umeclidinium/vilanterol (UMEC/VI) did not show clinically relevant effects on QT interval at clinical doses of FF, UMEC and VI.
No clinically relevant effects on the QTc interval were observed on review of centrally read ECGs from 911 subjects with COPD exposed to fluticasone furoate/umeclidinium/vilanterol for up to 24 weeks, or in the subset of 210 subjects exposed for up to 52 weeks.
Clinical efficacy and safety: The efficacy of TRELEGY ELLIPTA (92/55/22 micrograms), administered as a once-daily treatment, has been evaluated in patients with a clinical diagnosis of COPD in two, active-controlled studies and in a single, non-inferiority study. All three studies were multicentre, randomised, double-blind studies that required patients to be symptomatic with a COPD Assessment Test (CAT) score ≥10 and on daily maintenance treatment for their COPD for at least three months prior to study entry.
FULFIL (CTT116853) was a 24-week study (N=1,810), with an extension up to 52 weeks in a subset of subjects (n=430), that compared TRELEGY ELLIPTA (92/55/22 micrograms) with budesonide/formoterol 400/12 micrograms (BUD/FOR) administered twice-daily. At screening, the mean post-bronchodilator percent predicted FEV1 was 45% and 65% of patients reported a history of one or more moderate/severe exacerbation in the past year.
IMPACT (CTT116855) was a 52-week study (N=10,355) that compared TRELEGY ELLIPTA (92/55/22 micrograms) with fluticasone furoate/vilanterol 92/22 micrograms (FF/VI) and umeclidinium/vilanterol 55/22 micrograms (UMEC/VI). At screening, the mean post-bronchodilator percent predicted FEV1 was 46% and over 99% of patients reported a history of one or more moderate/severe exacerbation in the past year.
At study entry, the most common COPD medications reported in the FULFIL and IMPACT studies were ICS +LABA+LAMA (28%, 34% respectively), ICS+LABA (29%, 26% respectively), LAMA+LABA (10%, 8% respectively) and LAMA (9%, 7% respectively). These patients may have also been taking other COPD medications (e.g. mucolytics or leukotriene receptor antagonists).
Study 200812 was a 24-week, non-inferiority study (N=1,055) that compared TRELEGY ELLIPTA (92/55/22 micrograms) with FF/VI (92/22 micrograms) + UMEC (55 micrograms), co-administered once daily as a multi-inhaler therapy in patients with a history of moderate or severe exacerbations within the prior 12 months.
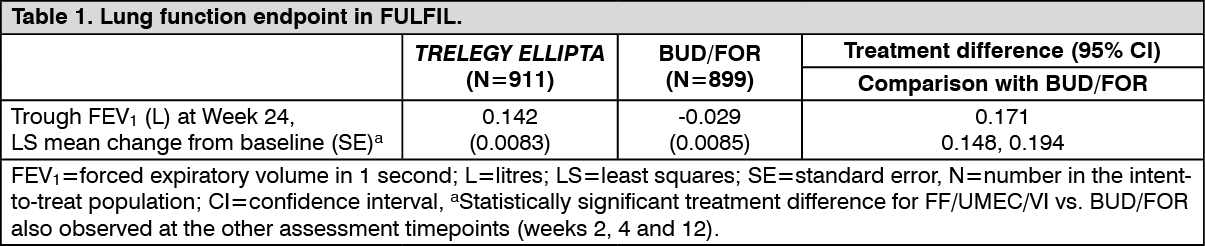
Lung Function: In FULFIL, bronchodilatory effects with TRELEGY ELLIPTA were evident on the first day of treatment and were maintained over the 24-week treatment period (mean changes from baseline in FEV1 were 90-222 mL on day 1 and 160-339 mL at week 24). TRELEGY ELLIPTA significantly improved (p<0.001) lung function (as defined by mean change from baseline in trough FEV1 at week 24) (see Table 1) and the improvement was maintained in the subset of patients who continued treatment to week 52. (See Table 1.)
 Click on icon to see table/diagram/image
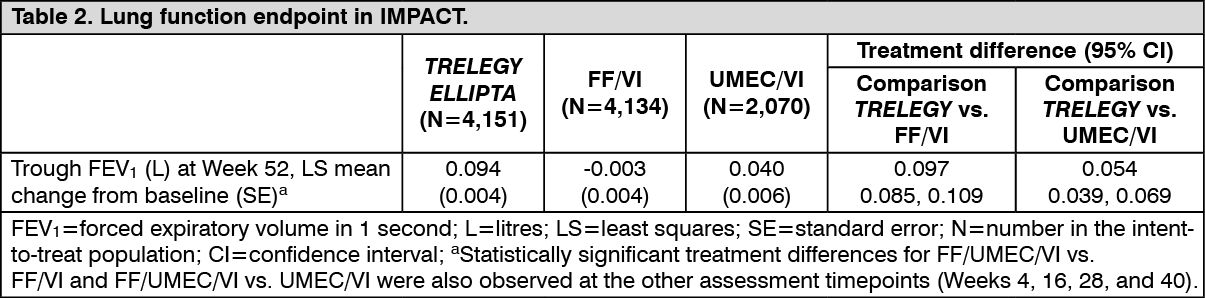
Click on icon to see table/diagram/imageIn IMPACT, TRELEGY ELLIPTA significantly improved (p<0.001) lung function when compared with FF/VI and UMEC/VI over a 52-week period (see Table 2).
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageIn Study 200812, TRELEGY ELLIPTA was non-inferior compared with FF/VI+UMEC, co-administered in two inhalers, in the improvement from baseline in trough FEV1 at Week 24. The pre-specified non-inferiority margin was 50 mL.
Exacerbations: In IMPACT, over 52 weeks, TRELEGY ELLIPTA significantly reduced (p<0.001) the annual rate of moderate/severe exacerbations by 15% (95% CI:10, 20) compared with FF/VI (rate; 0.91 vs 1.07 events per patient year) and by 25% (95% CI: 19, 30) compared with UMEC/VI (rate; 0.91 vs 1.21 events per patient year). In FULFIL, based upon data up to 24 weeks, TRELEGY ELLIPTA significantly reduced (p=0.002) the annual rate of moderate/severe exacerbations by 35% (95% CI: 14, 51) compared with BUD/FOR.
In IMPACT, TRELEGY ELLIPTA prolonged the time to first moderate/severe exacerbation and significantly decreased (p<0.001) the risk of a moderate/severe exacerbation, as measured by time to first exacerbation, compared with both FF/VI (14.8%; 95% CI: 9.3, 19.9) and UMEC/VI (16.0%; 95% CI: 9.4, 22.1). In FULFIL, TRELEGY ELLIPTA significantly decreased the risk of a moderate/severe exacerbation compared with BUD/FOR over 24 weeks (33%; 95% CI: 12, 48; p=0.004).
In IMPACT, treatment with TRELEGY ELLIPTA reduced the annual rate of severe exacerbations (i.e., requiring hospitalisation or resulting in death) by 13% compared with FF/VI (95% CI: -1, 24; p=0.064). Treatment with TRELEGY ELLIPTA significantly reduced the annual rate of severe exacerbations by 34% compared with UMEC/VI (95% CI: 22, 44; p<0.001).
Health-Related Quality of Life: TRELEGY ELLIPTA significantly improved (p<0.001) Health Related Quality of Life (as measured by the St George's Respiratory Questionnaire [SGRQ] total score) in both FULFIL (Week 24) when compared with BUD/FOR (-2.2 units; 95% CI: -3.5, -1.0) and IMPACT (Week 52) when compared with FF/VI (-1.8 units; 95% CI: -2.4, -1.1) and UMEC/VI (-1.8 units; 95% CI: -2.6, -1.0).
A higher percentage of patients receiving TRELEGY ELLIPTA responded with a clinically meaningful improvement in SGRQ total score in FULFIL at Week 24 compared with BUD/FOR (50% and 41% respectively), odds ratios of response vs. non-response (OR) (1.41; 95% CI: 1.16, 1.70) and in IMPACT at Week 52 compared with FF/VI and UMEC/VI (42%, 34% and 34% respectively), OR vs. FF/VI (1.41; 95% CI:1.29, 1.55) and OR vs. UMEC/VI (1.41; 95% CI: 1.26, 1.57); all treatment comparisons were statistically significant (p<0.001).
In FULFIL, the proportion of patients who were CAT responders (defined as 2 units below baseline or lower) at Week 24, was significantly higher (p<0.001) for patients treated with TRELEGY ELLIPTA compared with BUD/FOR (53% vs. 45%; OR 1.44; 95% CI: 1.19, 1.75). In IMPACT, the proportion of patients who were CAT responders at Week 52 was significantly higher (p<0.001) for patients treated with TRELEGY ELLIPTA (42%) compared with FF/VI (37%; OR 1.24; 95% CI: 1.14, 1.36) and UMEC/VI (36%; OR 1.28; 95% CI: 1.15, 1.43).
Symptom Relief: Breathlessness was measured using the Transition Dyspnoea Index (TDI) focal score at Week 24 in FULFIL and Week 52 in IMPACT (a subset of patients, n=5,058). In FULFIL the proportion of responders according to TDI (defined as at least 1 unit) was significantly higher (p<0.001) for TRELEGY ELLIPTA compared with BUD/FOR (61% vs 51%; OR 1.61; 95% CI: 1.33, 1.95). In IMPACT, the proportion of responders was also significantly higher (p<0.001) for TRELEGY ELLIPTA (36%) compared with FF/VI (29%; OR 1.36; 95% CI: 1.19, 1.55) and UMEC/VI (30%; OR 1.33; 95% CI: 1.13, 1.57).
In FULFIL, TRELEGY ELLIPTA improved daily symptoms of COPD as assessed by E-RS: COPD total score, compared with BUD/FOR (≥2 unit decrease from baseline). The proportion of responders during Weeks 21-24 was significantly higher (p<0.001) for patients treated with TRELEGY ELLIPTA compared with BUD/FOR (47% and 37% respectively; OR 1.59; 95% CI: 1.30, 1.94).
Use of Rescue Medication: In FULFIL, TRELEGY ELLIPTA significantly reduced (p<0.001) the use of rescue medication between Weeks 1-24 compared with BUD/FOR (treatment difference: -0.2 occasions per day; 95% CI: -0.3, -0.1).
In IMPACT, TRELEGY ELLIPTA significantly reduced (p<0.001) the use of rescue medication (occasions per day) at each 4-week time period compared with FF/VI and UMEC/VI. At Weeks 49-52, the treatment difference was -0.28 (95% CI: -0.37, -0.19) when compared with FF/VI and -0.30 (95% CI: -0.41, -0.19) with UMEC/VI.
Nighttime awakenings: In IMPACT, TRELEGY ELLIPTA statistically significantly reduced the mean number of nighttime awakenings due to COPD compared with FF/VI (-0.05; 95% CI: -0.08, -0.01; p=0.005) and with UMEC/VI (-0.10; 95% CI: -0.14, -0.05; p<0.001) at Weeks 49 to 52. Significant reductions were observed over all other timepoints for UMEC/VI (p<0.001) and for the all but two of the of timepoints for FF/VI (p≤0.021).
Pharmacokinetics: When fluticasone furoate, umeclidinium and vilanterol were administered in combination by the inhaled route from a single inhaler in healthy subjects, the pharmacokinetics of each component were similar to those observed when each active substance was administered either as fluticasone furoate/vilanterol combination or as an umeclidinium/vilanterol combination or umeclidinium monotherapy.
Population PK analyses for FF/UMEC/VI were conducted using a combined PK dataset from three phase III studies in 821 COPD subjects. Systemic drug levels (steady state Cmax and AUC) of FF, UMEC and VI following FF/UMEC/VI in one inhaler (triple combination) were within the range of those observed following FF/VI + UMEC as two inhalers, dual combinations (FF/VI and UMEC/VI), as well as individual 14 single inhalers (FF, UMEC and VI). Covariate analysis showed higher FF apparent clearance (42%) when comparing FF/VI to FF/UMEC/VI; however, this is not considered clinically relevant.
Absorption: Fluticasone furoate: Following inhaled administration of fluticasone furoate/umeclidinium/vilanterol in healthy subjects, fluticasone furoate Cmax occurred at 15 minutes. The absolute bioavailability of fluticasone furoate when administrated as fluticasone furoate/vilanterol by inhalation was 15.2%, primarily due to absorption of the inhaled portion of the dose delivered to the lung, with negligible contribution from oral absorption. Following repeat dosing of inhaled fluticasone furoate/vilanterol, steady state was achieved within 6 days with up to 1.6-fold accumulation.
Umeclidinium: Following inhaled administration of fluticasone furoate/umeclidinium/vilanterol in healthy subjects, umeclidinium Cmax occurred at 5 minutes. The absolute bioavailability of inhaled umeclidinium was on average 13%, with negligible contribution from oral absorption. Following repeat dosing of inhaled umeclidinium, steady state was achieved within 7 to 10 days with 1.5 to 2-fold accumulation.
Vilanterol: Following inhaled administration of fluticasone furoate/umeclidinium/vilanterol in healthy subjects, vilanterol Cmax occurred at 7 minutes. The absolute bioavailability of inhaled vilanterol was 27%, with negligible contribution from oral absorption. Following repeat dosing of inhaled umeclidinium/vilanterol, steady state was achieved within 6 days with up to 1.5-fold accumulation.
Distribution: Fluticasone furoate: Following intravenous dosing of fluticasone furoate to healthy volunteers, the mean volume of distribution at steady state of 661 litres. Fluticasone furoate has a low association with red blood cells. In vitro plasma protein binding in human plasma of fluticasone furoate was high, on average >99.6%.
Umeclidinium: Following intravenous administration of umeclidinium to healthy volunteers, the mean volume of distribution was 86 litres. In vitro plasma protein binding in human plasma was on average 89%.
Vilanterol: Following intravenous administration of vilanterol to healthy volunteers, the mean volume of distribution at steady state was 165 litres. Vilanterol has a low association with red blood cells. In vitro plasma protein binding in human plasma was on average 94%.
Biotransformation: Fluticasone furoate: In vitro studies showed that fluticasone furoate is primarily metabolised by cytochrome P450 3A4 (CYP3A4) and is a substrate for the P-gp transporter. The primary metabolic route for fluticasone furoate is hydrolysis of the S-fluoromethyl carbothioate group to metabolites with significantly reduced corticosteroid activity. Systemic exposure to the metabolites is low.
Umeclidinium: In vitro studies showed that umeclidinium is primarily metabolised by cytochrome P450 2D6 (CYP2D6) and is a substrate for the P-gp transporter. The primary metabolic routes for umeclidinium are oxidative (hydroxylation, O-dealkylation) followed by conjugation (glucuronidation, etc), resulting in a range of metabolites with either reduced pharmacological activity or for which the pharmacological activity has not been established. Systemic exposure to the metabolites is low.
Vilanterol: In vitro studies showed that vilanterol is primarily metabolised by cytochrome P450 3A4 (CYP3A4) and is a substrate for the P-gp transporter. The primary metabolic routes for vilanterol are O-dealkylation to a range of metabolites with significantly reduced beta1- and beta2-adrenergic agonist activity. Plasma metabolic profiles following oral administration of vilanterol in a human radiolabel study were consistent with high first-pass metabolism. Systemic exposure to the metabolites is low.
Elimination: Fluticasone furoate: The apparent plasma elimination half-life of fluticasone furoate following inhaled administration of fluticasone furoate/vilanterol was, on average, 24 hours. Following intravenous administration, the elimination phase half-life averaged 15.1 hours. Plasma clearance following intravenous administration was 65.4 litres/hour. Urinary excretion accounted for approximately 2% of the intravenously administered dose. Following oral administration, fluticasone furoate was eliminated in humans mainly by metabolism with metabolites being excreted almost exclusively in faeces, with <1% of the recovered radioactive dose eliminated in the urine.
Umeclidinium: Umeclidinium plasma elimination half-life following inhaled dosing for 10 days averaged 19 hours, with 3% to 4% active substance excreted unchanged in urine at steady-state. Plasma clearance following intravenous administration was 151 litres/hour. Following intravenous administration, approximately 58% of the administered radiolabelled dose was excreted in faeces and approximately 22% of the administered radiolabelled dose was excreted in urine. The excretion of the drug-related material in the faeces following intravenous dosing indicated secretion into the bile. Following oral administration, 92% of the administered radiolabelled dose was excreted primarily in faeces. Less than 1% of the orally administered dose (1% of recovered radioactivity) was excreted in urine, suggesting negligible absorption following oral administration.
Vilanterol: Vilanterol plasma elimination half-life following inhaled dosing for 10 days averaged 11 hours. Plasma clearance of vilanterol following intravenous administration was 108 litres/hour. Following oral administration of radiolabelled vilanterol, 70% of the radiolabel was excreted in urine and 30% in faeces. Primary elimination of vilanterol was by metabolism followed by excretion of metabolites in urine and faeces.
Special populations: Elderly: The effects of age on the pharmacokinetics of fluticasone furoate, umeclidinium and vilanterol were evaluated in the population pharmacokinetic analysis. No clinically relevant effects requiring dose adjustment were observed.
Renal impairment: The effect of fluticasone furoate/umeclidinium/vilanterol has not been evaluated in subjects with renal impairment. However, studies have been conducted with fluticasone furoate/vilanterol and umeclidinium/vilanterol that showed no evidence of an increase in systemic exposure to fluticasone furoate, umeclidinium or vilanterol. In vitro protein binding studies between subjects with severe renal impairment and healthy volunteers were conducted, and no clinically significant evidence of altered protein binding was seen.
The effects of haemodialysis have not been studied.
Hepatic impairment: The effect of fluticasone furoate/umeclidinium/vilanterol has not been evaluated in subjects with hepatic impairment. However, studies have been conducted with fluticasone furoate/vilanterol and umeclidinium/vilanterol.
The fluticasone furoate/vilanterol component of TRELEGY ELLIPTA was assessed in patients with all severities of hepatic impairment (Child-Pugh A, B or C). For fluticasone furoate, patients with moderate hepatic impairment showed up to three times higher systemic exposure (FF 184 micrograms); therefore, patients with severe hepatic impairment received half the dose (FF 92 micrograms). At this dose, no effects on systemic exposure were observed. Therefore, caution is advised in moderate to severe hepatic impairment, but no specific dose adjustment based on hepatic function is recommended. There was no significant increase in systemic exposure to vilanterol.
Patients with moderate hepatic impairment showed no evidence of an increase in systemic exposure to either umeclidinium or vilanterol (Cmax and AUC). Umeclidinium has not been evaluated in patients with severe hepatic impairment.
Other special populations: The effects of race, gender and weight on the pharmacokinetics of fluticasone furoate, umeclidinium and vilanterol were also evaluated in the population pharmacokinetic analysis.
In 113 East Asian subjects with COPD (Japanese and East Asian Heritage), who received FF/UMEC/VI from a single inhaler (27% subjects), fluticasone furoate AUC(ss) estimates were on average 30% higher compared with Caucasian subjects. However, these higher systemic exposures remain below the threshold for FF-induced reduction of serum and urine cortisol and are not considered clinically relevant. There was no effect of race on pharmacokinetic parameters of umeclidinium or vilanterol in subjects with COPD.
No clinically relevant differences requiring dose adjustment based on race, gender or weight were observed in fluticasone furoate, umeclidinium or vilanterol systemic exposure.
In terms of other patient characteristics, a study in CYP2D6 poor metabolisers showed no evidence of a clinically significant effect of CYP2D6 genetic polymorphism on systemic exposure to umeclidinium.
Toxicology: Pre-clinical safety data: Pharmacological and toxicological effects seen with fluticasone furoate, umeclidinium or vilanterol in non-clinical studies were those typically associated with glucocorticoids, muscarinic receptor antagonists, or beta2-adrenergic receptor agonists. Administration of combined fluticasone furoate, umeclidinium and vilanterol to dogs did not result in any significant new toxicity or any major exacerbation of expected findings associated with fluticasone furoate, umeclidinium or vilanterol alone.
Genotoxicity and carcinogenicity: Fluticasone furoate: Fluticasone furoate was not genotoxic in a standard battery of studies and was not carcinogenic in lifetime inhalation studies in rats or mice at exposures of 1.4- or 2.9-fold, respectively, those seen in humans at a daily dose of 92 micrograms fluticasone furoate, based on AUC.
Umeclidinium: Umeclidinium was not genotoxic in a standard battery of studies and was not carcinogenic in lifetime inhalation studies in mice or rats at exposures ≥20- or ≥17-fold the human clinical exposure at a daily dose of 55 micrograms umeclidinium, based on AUC respectively.
Vilanterol: Vilanterol (as alpha-phenylcinnamate) and triphenylacetic acid were not genotoxic indicating that vilanterol (as trifenatate) does not represent a genotoxic hazard to humans. Consistent with findings for other beta2 agonists, in lifetime inhalation studies vilanterol trifenatate caused proliferative effects in the female rat and mouse reproductive tract and rat pituitary gland. There was no increase in tumour incidence in rats or mice at exposures 0.9- or 22-fold, respectively, the human clinical exposure of vilanterol at a daily dose of 22 micrograms based on AUC.
Toxicity to reproduction: Fluticasone furoate, umeclidinium and vilanterol did not have any adverse effects on male or female fertility in rats.
Fluticasone furoate: Fluticasone furoate was not teratogenic in rats or rabbits, but delayed development in rats and caused abortion in rabbits at maternally toxic doses. There were no effects on development in rats at exposures 6.6-fold the human clinical exposure at a daily dose of 92 micrograms, based on AUC. Fluticasone furoate had no adverse effect on pre-or post-natal development in rats.
Umeclidinium: Umeclidinium was not teratogenic in rats or rabbits. In a pre- and post-natal study, subcutaneous administration of umeclidinium to rats resulted in lower maternal body weight gain and food consumption and slightly decreased pre-weaning pup body weights in dams given 180 micrograms/kg/day dose (approximately 61-fold the human clinical exposure of umeclidinium at a daily dose of 55 micrograms, based on AUC).
Vilanterol: Vilanterol was not teratogenic in rats. In inhalation studies in rabbits, vilanterol caused effects similar to those seen with other beta2-adrenergic agonists (cleft palate, open eyelids, sternebral fusion and limb flexure/malrotation). When given subcutaneously there were no effects at exposures 62-fold the human clinical exposure at a daily dose of 22 micrograms, based on AUC. Vilanterol had no adverse effect on pre- or post-natal development in rats.